MAFFT version 7
% mafft --add new_sequences --reorder existing_alignment > output
- Sequences in new_sequences are ungapped and then aligned to existing_alignment.
- new_sequences is a single multi-FASTA format file.
- existing_alignment is a single multi-FASTA format file.
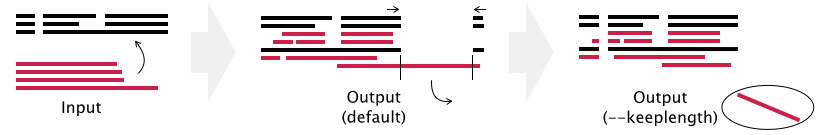
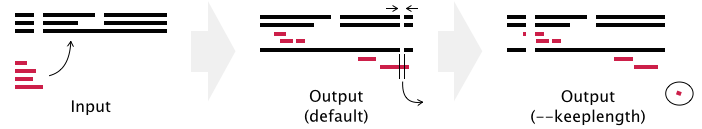
- Gaps in existing_alignment are preserved, but the alignment length may be changed in the default setting (see example below).
- If the --keeplength option is given, then the alignment length is unchanged. Insertions at the new sequences are deleted.
- Add --mapout to see a correspondence table of positions, new_sequences.map, between before and after the calculation. The --mapout option automatically turns on the --keeplength option, to keep the numbering of sites in the reference alignment (explanation added, 2016/Aug).
- Omit --reorder to preserve the original sequence order.
- Available in versions ≥6.811 (?); first described in Katoh & Frith 2012
- In versions ≥7.370, iterative refinement is performed if the --maxiterate n flag is given, where n > 0. The combination of iterative refinement and --keeplength is not supported (2017/Dec). New
- Accurate distance calculation (--localpair, --globalpair and --genafpair) can be applied to up to a few hundreds of sequences (2017/Dec). Updated
- Online version
- The difference between --addfull and --add will be explained later.
% mafft --auto --addfragments fragments --reorder --thread -1 existing_alignment > output
- Sequences in fragments are ungapped and then aligned to existing_alignment.
- fragments is a single multi-FASTA format file.
- existing_alignment is a single multi-FASTA format file.
- Gaps in existing_alignment are preserved, but the alignment length may be changed in the default setting (see example below).
- If the --keeplength option is given, then the alignment length is unchanged. Insertions at the fragmentary sequences are deleted.
- Add --mapout to see a correspondence table of positions, fragments.map, between before and after the calculation. The --mapout option automatically turns on the --keeplength option, to keep the numbering of sites in the reference alignment (explanation added, 2016/Aug).
- --auto automatically switches algorithm according to data size. Safer to always use this flag. (added 2020/Sep)
- --multipair uses a high-cost (in time and memory usage) option. Same as default. Applicable to ∼<30,000 sites × ∼<1,000 sequences.
- --6merpair uses a low-cost option.
- Omit --reorder to preserve the original sequence order.
- Described in Katoh & Frith 2012
- Can be used off-label to align closely-related sequences to a reference to build an MSA.
- Online version
Example
When existing_alignment is>seq1 ACCDEFGHI-K >seq2 A--DEFGHI-Kand a sequence to be added (newseq) is>newseq ACCDPQRSTEFGthen the result of mafft --addfull (and mafft --addfragments, mafft --add) is% mafft --addfull newseq existing_alignment seq1 ACCD-----EFGHIK seq2 A--D-----EFGHIK newseq ACCDPQRSTEFG--- * * ***The alignment length is changed (11→15) in this case, as PQRST is inserted (+5) and a gap-only column (between I and K) is removed (-1).With the --keeplength option, the insertion PQRST is removed and the alignment length is kept unchanged (11→11).
% mafft --addfull newseq --keeplength existing_alignment seq1 ACCDEFGHI-K seq2 A--DEFGHI-K newseq ACCDEFG---- * ****With the --mapout option, a correspondence table of positions is output to the newseq.map file.
% mafft --addfull newseq --mapout existing_alignment % cat newseq.map >newseq # letter, position in the original sequence, position in the reference alignment A, 1, 1 C, 2, 2 C, 3, 3 D, 4, 4 P, 5, - Q, 6, - R, 7, - S, 8, - T, 9, - E, 10, 5 F, 11, 6 G, 12, 7The --compactmapout option outputs the same information in a more compact form. Available in versions 7.496 and higher. 2021/Dec
% mafft --addfull newseq --compactmapout existing_alignment % cat newseq.map # Insertions in the added sequences > Position in reference >newseq 5P - 9T > 4v5The --mapout and --compactmapout options automatically turn on the --keeplength option, to keep the consistency of the numbering of positions in the reference.
Possible misalignment by versions ≤6.815:existing alignment: AAAAAAAA---------BBBBBBBBB existing alignment: AAAAAAAA---------BBBBBBBBB existing alignment: AAAAAAAA------------------ new sequence: AAAAAAAABBBBBBBBB---------Fixed in versions ≥6.817:
existing alignment: AAAAAAAABBBBBBBBB existing alignment: AAAAAAAABBBBBBBBB existing alignment: AAAAAAAA--------- new sequence: AAAAAAAABBBBBBBBB
% mafft --addprofile aligned_sequences existing_alignment > output
- aligned_sequences are converted to a profile and then aligned to existing_alignment.
- aligned_sequences should be a single multi-FASTA format file.
- Both aligned_sequences and existing_alignment are preserved.
- existing_alignment should be another multi-FASTA format file.
- Accurate options (mafft-linsi, mafft-ginsi and mafft-einsi), instead of mafft, can be used for a few hundreds of sequences.
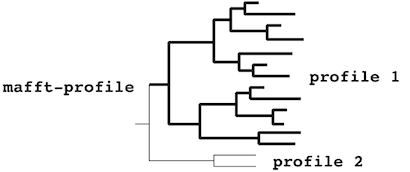
- aligned_sequences must form a monophyletic cluster, as they are converted into a single profile. Otherwise, the calculation stops.
- existing_alignment must form a paraphyletic cluster:
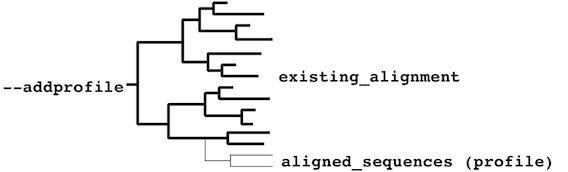
or a monophyletic cluster:
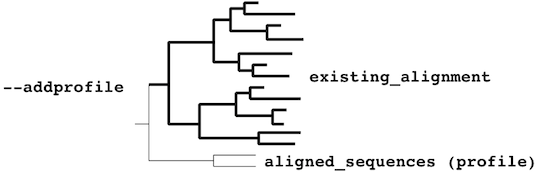
The mafft-profile program assumes that
each profile separately forms
a monophyletic cluster.