The --seed option
The
--seed option can be used for adding unaligned sequences into a highly reliable alignment (seed) consisting of a small number of sequences.
In this option, the
aligned letters in the seed alignment are preserved but
gaps are not necessarily preserved.
If the given alignment (including the gap pattern) has to be completely preserved,
use the --add or --addfragments option.
Sequence-to-skeleton alignment
% mafft-linsi --seed skeleton_alignment unaligned_sequences > output
is applicable to:
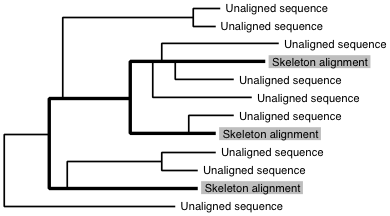
A full MSA is constructed while preserving the skeleton alignment.
If the number of unaligned sequences is much smaller than the number of sequences in the skeleton alignment,
use the --add option (supported in versions 6.8 and higher).
% mafft --add unaligned_sequences skeleton_alignment > output
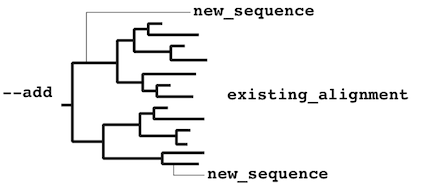
Sequence-to-group alignment
If you have version 6.8 or higher, use a faster option, --add.
The following information is obsolete.
% mafft-linsi --seed aligned_group new_sequence > output
is applicable to:
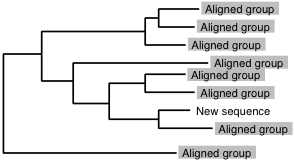
or
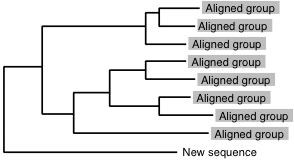 mafft-linsi
mafft-linsi can be replaced with other aliases. →
See the
algorithm
and
manual pages.
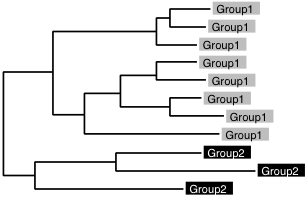
Group-to-group alignment
If you have version 6.8 or higher, use a faster option, --addprofile.
The following information is obsolete.
% mafft-linsi --seed group1 --seed group2 /dev/null > output
or
% mafft-profile group1 group2 > output
The former is more accurate but slower.
These are applicable to: