About
MAFFT
is a multiple sequence alignment program for
unix-like operating systems.
It offers a range of multiple alignment methods,
L-INS-i (accurate; recommended for <200 sequences),
FFT-NS-2 (fast; recommended for >2,000 sequences),
etc.
Merits
- (Accuracy)
L-INS-i is one of the most accurate multiple sequence alignment methods currently available.
L-INS-i is in particular suitable to align 10-100 protein sequences,
because of an objective function combining the WSP and consistency scores.
Protein benchmarks are in:
RNA benchmark:
DNA benchmark:
Phylogeny-based benchmark:
- (Scalability)
FFT-NS-2 and other progressive methods can align
many and/or long DNA/protein sequences, because of an FFT approximation
and a linear-space DP algorithm.
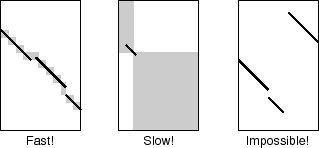
- The scoring system was designed to allow large gaps.
Thus MAFFT is suitable for LSU rRNA and SSU rRNA alignments that
sometimes have variable loop regions.
Staggered gaps (like the figure below) are also allowed.
This feature is remarkable with the --addfragments option.
Limitations
- (Accuracy)
Library extension is not
performed unlike
TCoffee,
because we think at present that iterative refinement is more efficient
than library extension.
- (Scalability)
If two unrelated and long genomic DNA sequences are given,
FFT-NS-2 tries to make a full-length alignment using
rigorous DP and requires large CPU time.
For such a case, homology search tools such as FASTA and BLAST are more suitable.
- The order of alignable blocks or domains are assumed to be conserved
for all input sequences.