Rapid calculation of full-length MSA of closely-related viral genomes Experimental (2020/Apr/11 )
When the input data set is large and the sequences are very closely related (% identity ∼ 99), it's sometimes useful to align all sequences just to a reference to build a full MSA.
Time complexity is
O(
N L log
L), where
N is the number of sequences and
L is sequence length.
Online version supports more than 20,000 sequences × ∼30,000 sites, 2021/Jan.
Online version supports up to ∼ 20,000 sequences × ∼30,000 sites, 2020/Apr/18.
Reduced the frequency of "timeout" error in data transfer, 2020/Jul/27.
On command line, use
version 7.467 or later.
Earlier versions (≤7.458) had the same options but were inefficient for this purpose.
Online
Input page Updated 2022/Mar
Procedure:
-
Select a single reference sequence or a reference MSA (a small set of sequences already aligned).
-
Input the reference (eg, NC_045512.2) to the "Reference" box.
-
Input the other sequences to the "New sequence(s)" box.
-
Select options (Adjust direction, Keep alignment length, etc) as necessary.
-
Submit
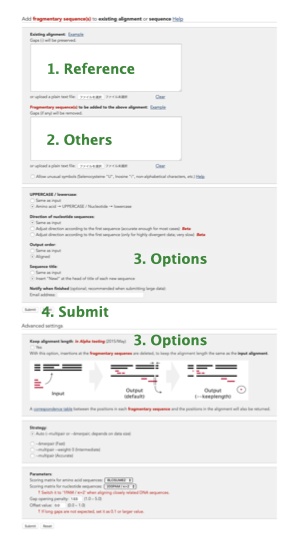
Note that:
-
The page title, addfragments, and labels of the two input boxes are not directly descriptive for this usage,
because this function is originally for another purpose.
-
If selecting the "Keep alignment length" option below in the input page, no gaps are inserted to the reference sequence, ie, corresponding sites in the other sequences are deleted.
As a result, the numbering of sites is kept and the calculation is faster than the default in some cases.
-
Don't change the "Strategy" switch from Auto when the sequences are long.
-
For less closely related sequences (% identity < ??), normal MSA calculation is probably necessary.
-
A similar option, addsequences, is not efficient for this purpose, but suitable when the input sequences are less closely related, the sequences to be added are fewer and a reference MSA is available.
-
Feedback.
Command Updated 2021/Jan/29
After dividing the input sequences into reference and others,
type:
% mafft --6merpair --addfragments othersequences referencesequence > output
To keep the numbering of sites,
% mafft --6merpair --keeplength --addfragments othersequences referencesequence > output
(sometimes faster than the default).
If the input data contains many ambiguous letters, try:
% mafft --6merpair --maxambiguous 0.05 --addfragments othersequences referencesequence > output
which removes sequences that have more than 5% ambiguous letters.
Available in versions ≥7.473.
Runs efficiently in parallel,
% mafft --6merpair --thread -1 --keeplength --addfragments othersequences referencesequence > output
Without the --6merpair flag, too slow for this purpose.
A similar option, --add, is not efficient for this purpose, but suitable when the input sequences are less closely related, the sequences to be added are fewer and a reference MSA is available.

