MAFFT version 7
Source package onlyVersions ≥7.350 support MPI. Linux only.
Two environmental variables, MAFFT_N_THREADS_PER_PROCESS and MAFFT_MPIRUN, have to be set.An example to use 160 cores (16 cores × 10 hosts):
The number of threads to run in a process: $ export MAFFT_N_THREADS_PER_PROCESS="1" Set "1" unless using a MPI/Pthreads hybrid mode. Location of mpirun/mpiexec and options: $ export MAFFT_MPIRUN="/somewhere/bin/mpirun -n 160 -npernode 16 -bind-to none ..." (for OpenMPI) $ export MAFFT_MPIRUN="/somewhere/bin/mpirun -n 160 -perhost 16 -binding none ..." (for MPICH) mpirun or mpiexec must be from the same library as mpicc that was used in compiling. Depending on the configuration of your cluster, LD_LIBRARY_PATH may be necessary to set. $ export LD_LIBRARY_PATH="/somewhere/lib" (Optional) Location of temporary directory (see below): $ export MAFFT_TMPDIR="/location/of/shared/filesystem/"To avoid typing these commands each time, try batch, in which parameters are easily set.
Add "--mpi --large" to the normal command of G-INS-1, L-INS-1 or E-INS-1.G-large-INS-1: $ mafft --mpi --large --globalpair --thread 16 input L-large-INS-1: $ mafft --mpi --large --localpair --thread 16 input E-large-INS-1: $ mafft --mpi --large --genafpair --thread 16 input E-large-INS-1 (old parameters): $ mafft --mpi --large --oldgenafpair --thread 16 inputThe --thread flag specifies the maximum number of threads (16 in these examples) used in step 2 (see below) and other calculations performed on a single node. It must be less than or equal to the number of physical cores in a single host. (Changed from --threadtb to --thread, 2018/Sep/22)
To set the environmental variables and run the command at a time, a batch script can be used.$ sh mpionly.noschedulerEdit the mpionly.noscheduler script according to your cluster's environment, and run it. Detailed information about the variables are explained in the batch file itself.For job schedulers, edit and run one of the following templates. If unsuccessful, try the script above without scheduler for small input sequence data, to identify the cause.
LSF:
$ bsub < mpionly.lsfMany cluster with the SGE/UGE scheduler have a parallel environment (PE) to use a specific number of cores per host. An example with a PE name mpi16:
$ qsub mpionly.ugeAsk system administrator if there is an appropriate PE.Otherwise, disable multithread.
$ qsub singlethread.ugeIn this case, step 2 runs in serial.PBS:
$ qsub mpionly.pbsSLURM:
preparing
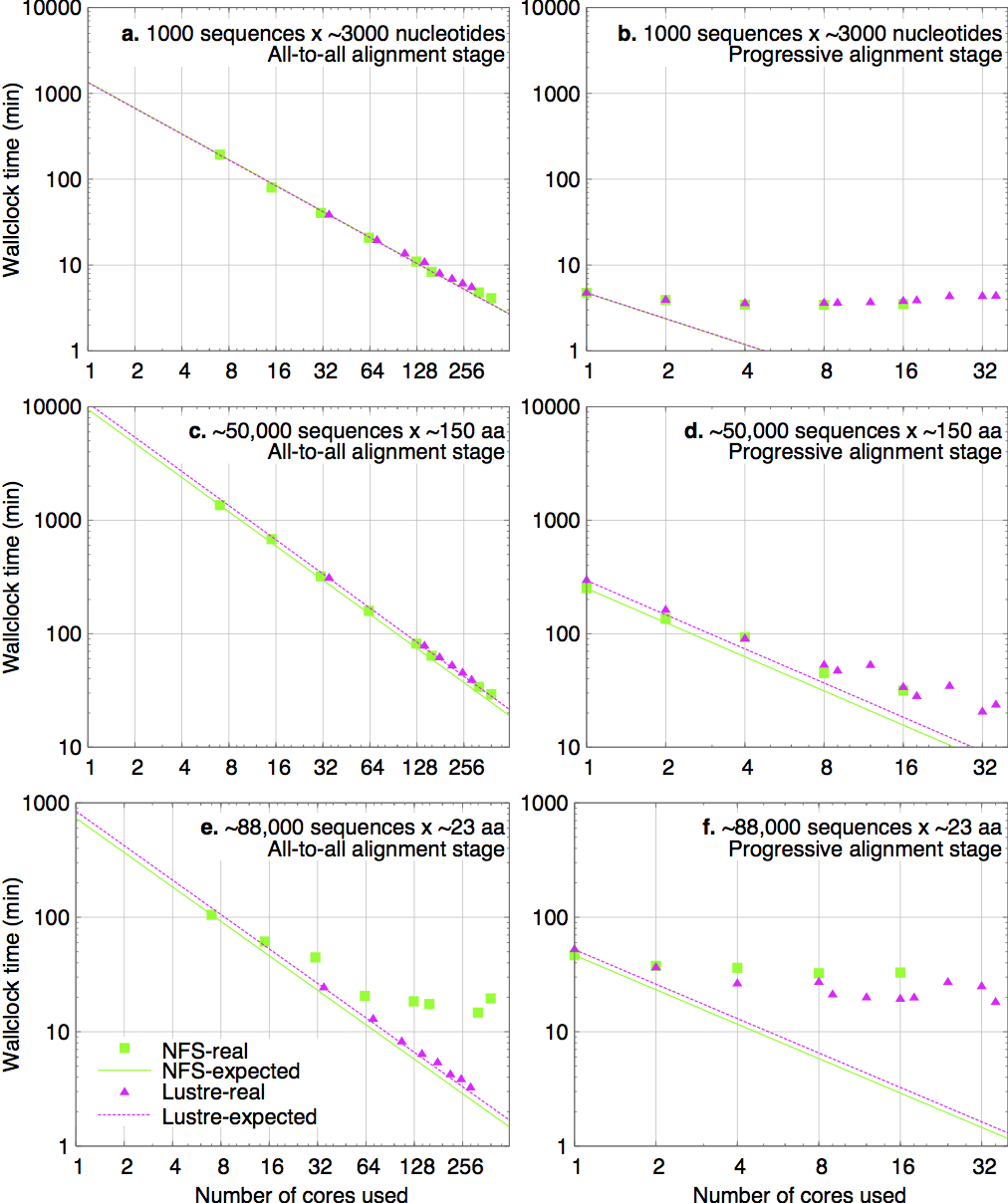
Wall-clock times in three cases are shown in the figure below. Panels a, c and e → step 1; Panels b, d and f → step 2.
% setenv MAFFT_TMPDIR /location/of/shared/filesystem/ (tcsh, csh, etc) $ export MAFFT_TMPDIR=/location/of/shared/filesystem/ (bash, zsh, etc)In the cases of the plots above, the efficiency with Lustre temporary directory is higher than that with NFS, especially in panel c (short sequences).
are welcome.