
MAFFT version 7
MAFFT outputs just the guide tree without alinging the sequences by:
% mafft --retree 0 --treeout input > outputThe resulting tree is put into the input.tree file in the Newick format.
% cat input.tree (((1_M63632:0.15750,2_U22180:0.15750):0.03300,(3_M92038 ...The sequences are numbered according to the order in the input file and the number is added to the each sequence name in the output tree.
If the number of input sequences is relatively small, <∼5,000, we can use a relatively accurate distance measure, which is based on all-to-all pairwise local or global alignments:
% mafft --retree 0 --treeout --globalpair --reorder input > output
% mafft --retree 0 --treeout --localpair --reorder input > outputThe former (--globalpair; uses global alignments) is expected to be suitable to compare sequences of similar lengths. The latter (--localpair; uses local alignments) is expected to be suitable to identify the relationship of truncated sequences and its full-length relative. However, the difference between them in the performance is not yet fully tested on actual data.
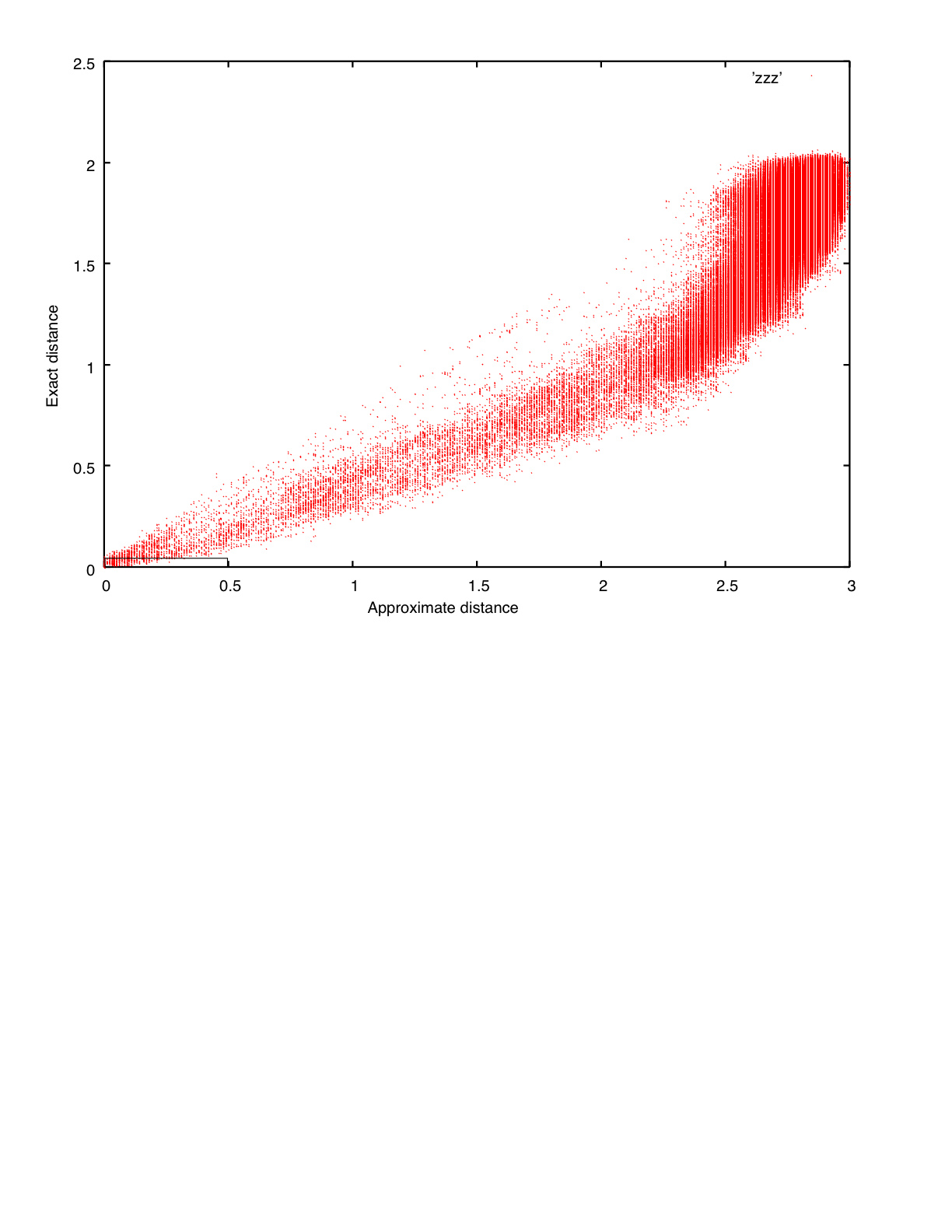
If the number of input sequences is relatively large, ∼5,000-∼50,000, a rough distance measure, 6mer distance: (Higgins & Sharp 1988; Jones et al. 1992; Katoh et al. 2002), is applicable.
% mafft --retree 0 --treeout --reorder input > output
The correlation between the two distances can be seen here.
For the above two cases, a tree-building method can be selected from
When the --reorder argument is given, the input sequences are re-ordered according to the similarity, but not aligned, and returned to standard output.
For further more sequences, ∼50,000-∼100,000, the PartTree algorithm can be applied:% mafft --retree 0 --treeout --parttree --reorder input > output
% mafft --retree 0 --treeout --dpparttree --reorder input > output
% mafft --retree 0 --treeout --fastaparttree --reorder input > output
When PartTree is applied, only a number is used to represent each sequence. The number (1, 2, ...) is the position of the sequence in the input file.
(((1,2),(3...
{kind=link}