
MAFFT version 6
Benchmark results of RNA aligners (May, 2008)
| Method | Accuracy of alignment | Accuracy of predicted secondary structure (MCC) | |||||||
| Base-pairing prob. | SPS | SCI | Pfold | McCaskill- MEA |
RNAalifold | (internal) | CPU time (s) | ||
| Structural alignment methods | |||||||||
| X-INS-i-scarnapair (Uses MXSCARNA) | McCaskill | 0.880 | 0.769 | 0.736 | 0.708 | 0.731 | - | 340 | |
| CONTRAfold | 0.882 | 0.776 | 0.735 | 0.706 | 0.728 | - | 510 | ||
| X-INS-i-foldalignglobalpair (Uses global alignments by FOLDALIGN) | McCaskill | 0.884 | 0.783 | 0.738 | 0.705 | 0.734 | - | 9,700 | |
| CONTRAfold | 0.884 | 0.785 | 0.735 | 0.707 | 0.738 | - | 10,000 | ||
| X-INS-i-foldalignlocalpair (Uses local alignments by FOLDALIGN) | McCaskill | 0.859 | 0.768 | 0.730 | 0.706 | 0.729 | - | 14,000 | |
| CONTRAfold | 0.875 | 0.780 | 0.725 | 0.706 | 0.734 | - | 14,000 | ||
| X-INS-i-larapair (Uses LaRA 1.3.2a) | McCaskill | 0.869 | 0.783 | 0.738 | 0.712 | 0.741 | - | (2,300) | |
| CONTRAfold | 0.871 | 0.788 | 0.741 | 0.709 | 0.745 | - | (2,500) | ||
| Q-INS-i | McCaskill | 0.877 | 0.741 | 0.730 | 0.701 | 0.695 | - | 54 | |
| CONTRAfold | 0.874 | 0.743 | 0.723 | 0.699 | 0.696 | - | 210 | ||
| RNA sampler | 0.809 | 0.789 | 0.733 | 0.700 | 0.725 | 0.699 | 6,900 | ||
| MASTR | 0.824 | 0.748 | 0.677 | 0.685 | 0.692 | 0.700 | 5,400 | ||
| LaRA 1.3.2a | 0.864 | 0.772 | 0.745 | 0.710 | 0.728 | - | (2,300) | ||
| Murlet | 0.875 | 0.737 | 0.702 | 0.705 | 0.705 | - | 4,800 | ||
| MXSCARNA 2 | 0.866 | 0.734 | 0.729 | 0.701 | 0.707 | - | 73 | ||
| StrAl | 0.809 | 0.699 | 0.662 | 0.662 | 0.675 | - | 18 | ||
| Sequence-based methods | |||||||||
| ProbConsRNA | 0.874 | 0.721 | 0.708 | 0.689 | 0.684 | - | 16 | ||
| G-INS-i | 0.866 | 0.719 | 0.710 | 0.684 | 0.681 | - | 3.5 | ||
| FFT-NS-2 | 0.832 | 0.674 | 0.678 | 0.663 | 0.669 | 1.2 | ←Default of mafft | ||
| ClustalW version 2 (Iteration=tree) | 0.798 | 0.641 | 0.649 | 0.641 | 0.652 | - | 22 | ||
| ClustalW version 2 (Default) | 0.795 | 0.646 | 0.640 | 0.641 | 0.648 | - | 2.6 | ||
A similar observation is made with the comparison between LaRA and X-INS-i-larapair. These two methods use the same pairwise structural alignment algorithm, LaRA.
The combination of X-INS-i and FOLDALIGN is slightly more accurate than the combination of X-INS-i and SCARNA. However, the latter is much faster than the former and the difference in the accuracy is quite small. From the practical viewpoint, we selected SCARNA as the default for X-INS-i.
The difference between global and local options of FOLDALIGN is probably because the dataset is globally alignable.
Several alignment methods (RNA sampler, Murlet, MXSCARNA and MASTR) return secondary structures internally predicted, while other methods (MAFFT, LaRA and StrAl) do not. At present, the advantage of internal prediction is not clear, since the accuracies of predictions by external methods (Pfold, McCaskill-MEA and RNAalifold) are comparable to or rather higher than those of internal predictions (shown in the '(internal)' column). This observation is consistent with the Murlet paper.
Each alignment by each method was subjected to three RNA secondary structure prediction programs, Pfold, McCakill-MEA and RNAalifold. Accuracies of the predicted structures were assessed with Matthews Correlation Coefficient (MCC), calculated from TP, TN, FP and FN by the compare_ct.pl program. The curated structures in the Rfam database were assumed as correct.
Various combinations of alignment programs and RNA secondary structure prediction programs were examined as in the figure below.
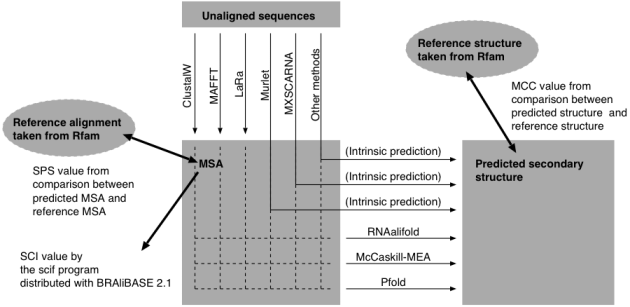
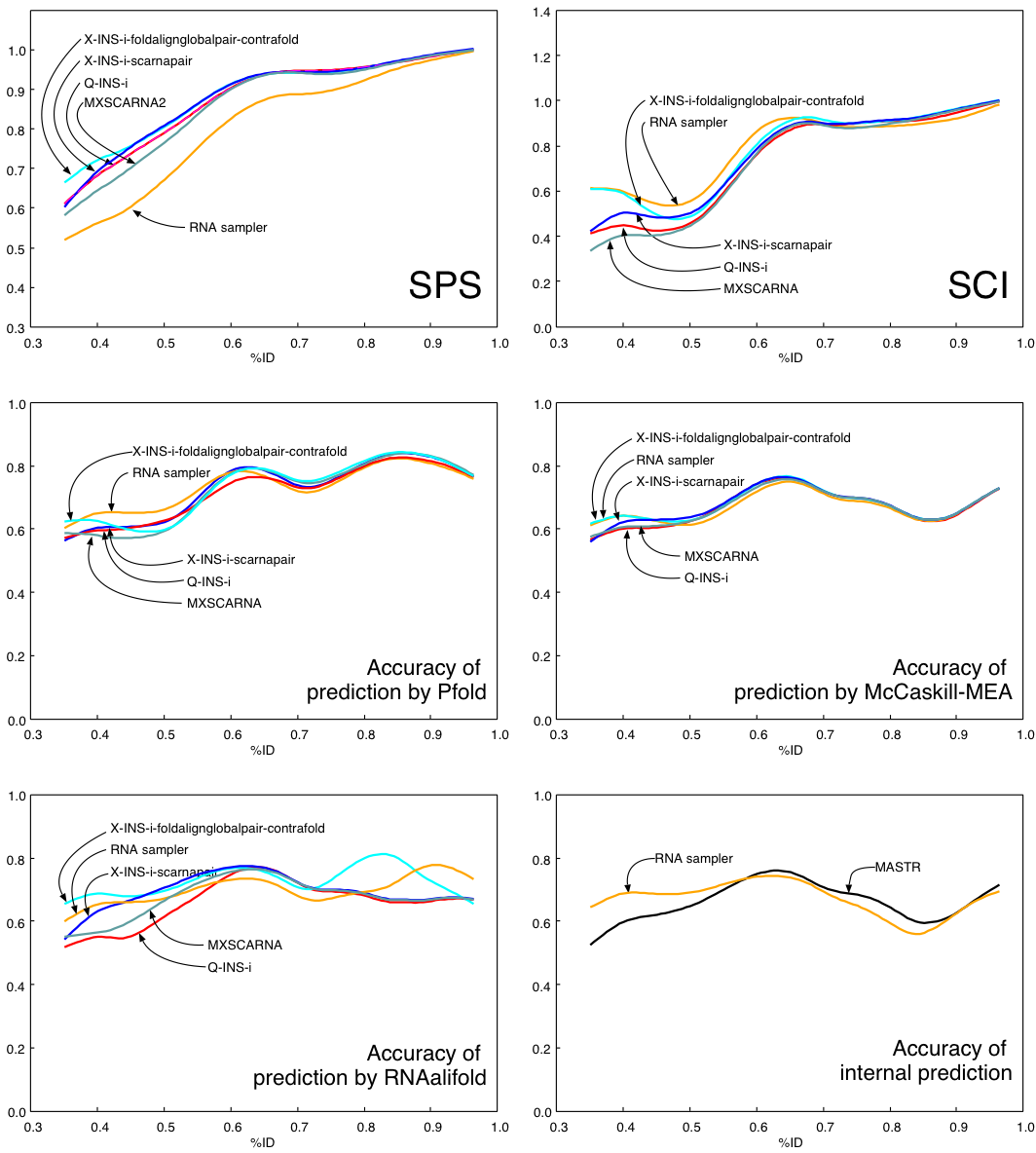
For each alignment method, the accuracy values of all of the 52 alignments were plotted as a function of average percent identity and then a curve was plotted using a cubic spline. The curves of RNA sampler (the best of existing methods) and MASTR (only for internal prediction) are also shown for reference.