 Bug information
Bug information (2014/Apr/08):
This feature was not compatible with version 7.130.
This problem has been fixed in version 7.147.
Re-aligning particular regions in a given MSA (in beta testing, 2013/Jul/12)
This is a semi-automatic alignment strategy.
Suppose that some sites are manually aligned based on solid biological evidence, but remaining sites are not aligned or just roughly aligned.
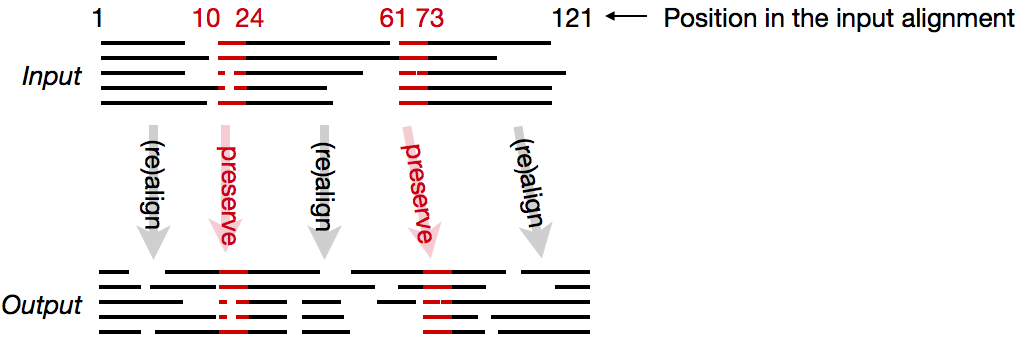
MAFFT can (re)align the latter sites while preserving the alignment(s) of former sites.
-
Download the regionalrealignment.rb script,
and edit line 3 according to where you installed mafft.
1 #! /usr/bin/env ruby
2
3 $MAFFTCOMMAND = '"/usr/local/bin/mafft"'
4 # Edit the above line to specify the location of mafft.
5 # $MAFFTCOMMAND = '"C:\folder name\mafft.bat"' # windows 7, 8
6 # $MAFFTCOMMAND = '"/usr/local/bin/mafft"' # mac, cygwin
7 # $MAFFTCOMMAND = '"/usr/bin/mafft"' # linux (rpm), ubuntu on windows 10 (deb)
8 # $MAFFTCOMMAND = '"/somewhere/mafft.bat"' # all-in-one version for linux or mac
Ruby is required.
-
Ruby is installed in MacOSX and most Linux systems.
-
On Windows 10, Ubuntu is useful for running Ruby and MAFFT.
-
Ruby installer for Windows 7, 8.
-
Create a setting file.
1 9 realign --maxiterate 100 # Comment can be added after #.
10 24 preserve # Preserve sites 10-24
25 60 realign --maxiterate 100 --tm 100 # Use transmembrane model for sites 25-60
61 73 preserve # Preserve sites 61-73
74 121 realign --maxiterate 100 --localpair # Realign sites 74-121
treeoption --localpair --thread -1 # "--thread -1" is applicable only on Linux and Mac.
-
The first column is the start position of each region.
-
The second column is the end position of each region.
-
If the third column is "realign", the region will be realigned.
-
If the third column is "preserved", the region will be preserved.
-
If command-line option for mafft is given after "realign", this option is used for aligning this region.
Different algorithms and parameters can be used for different regions.
-
The line starting with treeoption specifies the option to build a guide tree, which
is used for all the regions to be realigned.
-
If the number of sequences is large, --6merpair is recommended.
-
Otherwise, --localpair, --globalpair or --genafpair is recommended.
-
Run the regionalrealignment.rb script (edited in step 0).
% ruby regionalrealignment.rb setting input > output
-
The option used for each region is displayed to stderr at the end of calculation.
Tree: computed with --localpair --treeout
1 - 9: realigned with --maxiterate 100 --treein (tree)
10 - 24: preserved
25 - 60: realigned with --maxiterate 100 --tm 100 --treein (tree)
61 - 73: preserved
74 - 121: realigned with --maxiterate 100 --localpair --treein (tree)
This is just for confirmation.
