
MAFFT version 7
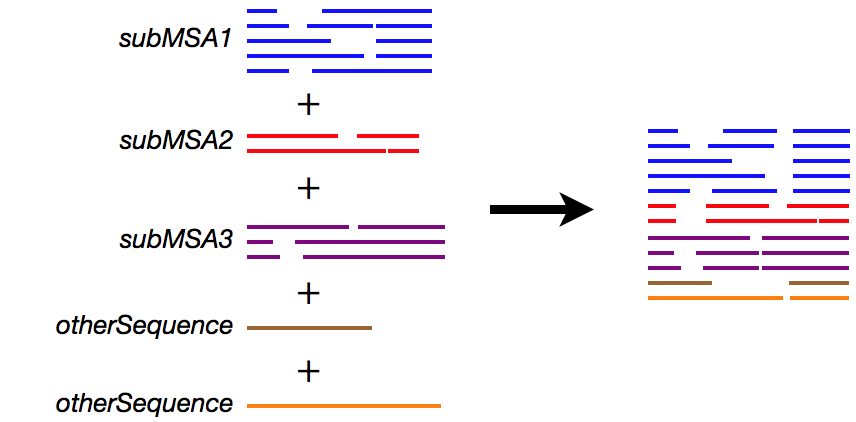
% cat subMSA1 subMSA2 subMSA3 otherSequences > input
Updated (2020/May/14) Note that each file must have "end of line" at the end of file. If not sure, type:
% echo >> subMSA1 % echo >> subMSA2 % echo >> subMSA3 % echo >> otherSequencesbefore running cat.
% ruby makemergetable.rb subMSA1 subMSA2 subMSA3 > subMSAtableThe subMSAtable file is:
1 2 3 4 5 # this is comment. subMSA1 6 7 8 # you can write anything after # 9 10 # subMSA3This means:
The subMSAtable file can be manually edited as necessary.
% mafft --merge subMSAtable input > output
A more rigorous distance measure can be used for small data.
% mafft --localpair --merge subMSAtable input > output
Iterative refinement is supported in version 7.040 and higher.
% mafft --localpair --maxiterate 100 --merge subMSAtable input > output
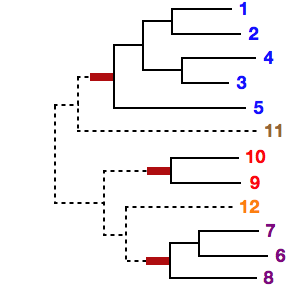
If phylogenetically unnatural grouping (eg, "1 5" in the above case) is given, the following warning appears and the calculation becomes slow.
# Sequences 1 and 2 seem to be closely related, but are not in the same sub MSA (1) in your setting.This warning has been changed in v7.236 (2015/Jun).
If inconsistent groupings (eg, "1 2" and "1 3") are given, the program stops.
By the --treein option, a user-defined guide tree can be given. In this case, each sub-MSA must form a monophyletic cluster in the given guide tree. Otherwise, the program stops.
% mafft --treein tree --merge subMSAtable input > output
The --merge option completely covers the situations where the mafft-profile program was used. So, mafft-profile will be deleted in the future.
% mafft-profile subMSA1 subMSA2 > output↓↓↓
% cat subMSA1 subMSA2 > input % ruby makemergetable.rb subMSA1 subMSA2 > subMSAtable % mafft --merge subMSAtable input > output
 Bug information (2013/May/26):
Bug information (2013/May/26):
In the multithread mode (--thread #), the --merge feature did not work in version 7.040.
This bug has been fixed in version 7.043.
% mafft --localpair --seed seed --merge subMSAtable input > outputThe sequences in the seed file are first numbered (1, 2, 3, ..), and the sequences in the input file are numbered (4, 5, 6, 7 ...). This numbering must be used in the subMSAtable and tree files.
To generate subMSAtable in this case, use the -s option to specify the number of sequences in the seed, eg, the seed file has three sequences,
% ruby makemergetable.rb -s 3 subMSA1 subMSA2 > subMSAtableThe resulting subMSAtable is:
4 5 6 7 8 # subMSA1 9 10 11 # subMSA2This can be used for the above command.